## number of barcodes: 4100 ## number of bins: 0 ## number of genes: 0 ## number of peaks: 0 ## number of motifs: 0
Step 2. Add cell-by-bin matrix
Next, we add the cell-by-bin matrix of 5kb resolution to the snap object. This function will automatically read the cell-by-bin matrix and add it to bmat slot of snap object.
# show what bin sizes exist in atac_v1_adult_brain_fresh_5k.snap file > showBinSizes("atac_v1_adult_brain_fresh_5k.snap"); [1] 1000500010000 > x.sp = addBmatToSnap(x.sp, bin.size=5000, num.cores=1);
Step 3. Matrix binarization
We will convert the cell-by-bin count matrix to a binary matrix. Some items in the count matrix have abnormally high coverage perhaps due to the alignment errors. Therefore, we next remove 0.1% items of the highest coverage in the count matrix and then convert the remaining non-zero items to 1.
> x.sp = makeBinary(x.sp, mat="bmat");
Step 4. Bin filtering
First, we filter out any bins overlapping with the ENCODE blacklist to prevent from potential artifacts.
## number of barcodes: 4100 ## number of bins: 545183 ## number of genes: 0 ## number of peaks: 0 ## number of motifs: 0
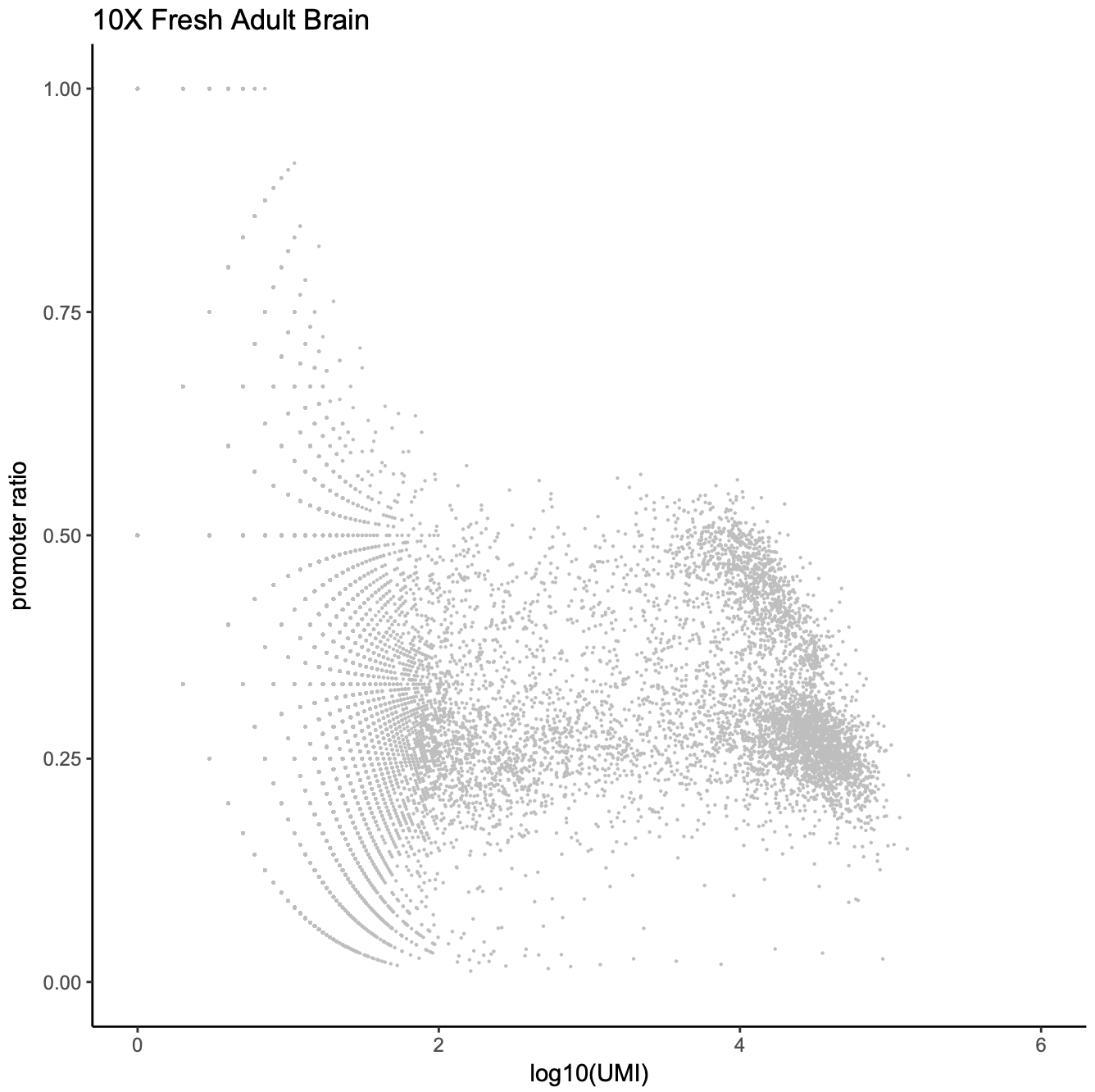
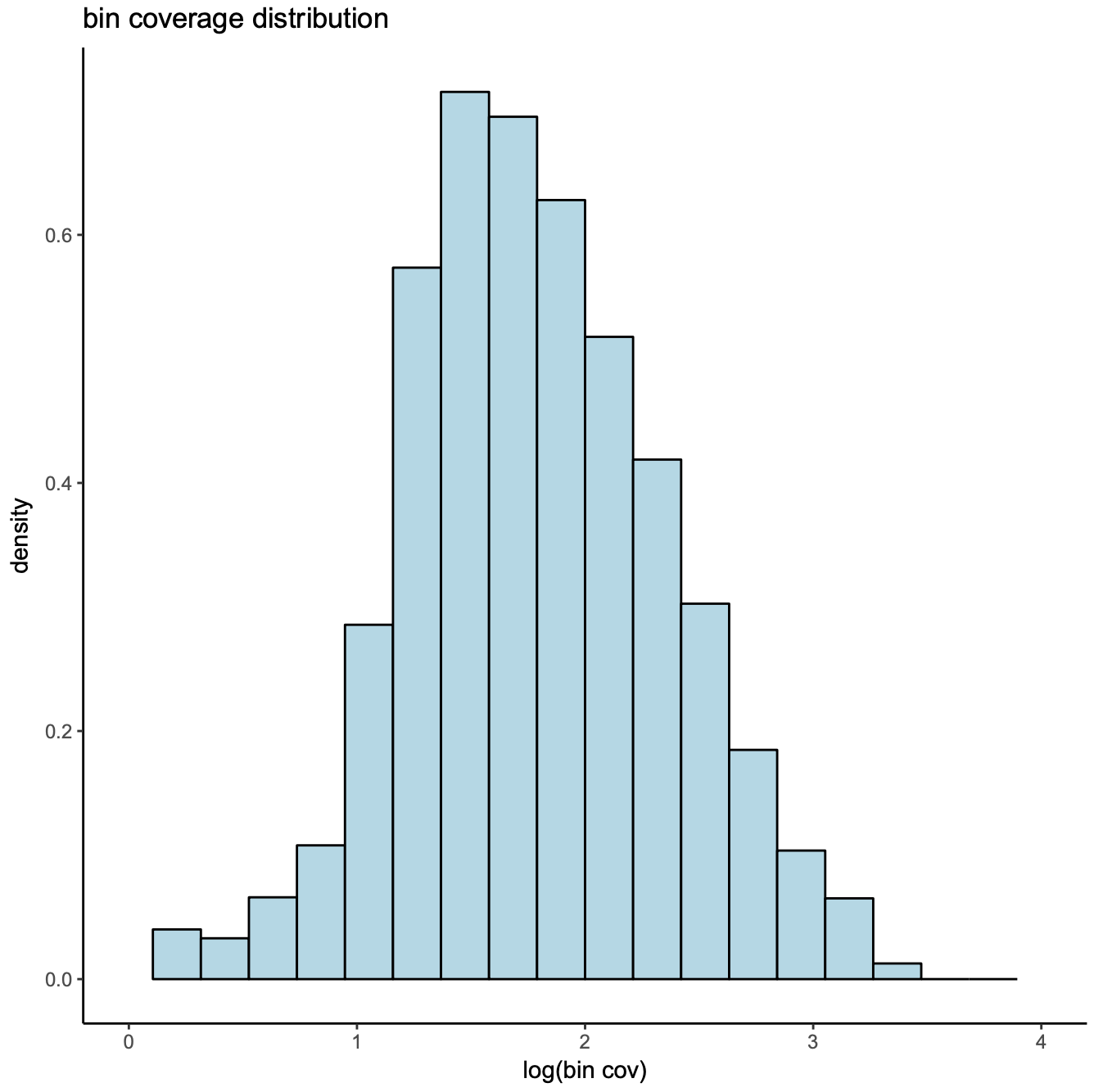
Third, the bin coverage roughly obeys a log normal distribution. We remove the top 5% bins that overlap with invariant features such as promoters of the house keeping genes.
We next determine the number of reduced dimensions to include for downstream analysis. We use an ad hoc method by simply looking at a pairwise plot and select the number of dimensions in which the scatter plot starts looking like a blob. In the below example, we choose the first 20 dimensions.
Using the selected significant dimensions, we next construct a K Nearest Neighbor (KNN) Graph (k=15). Each cell is a node and the k-nearest neighbors of each cell are identified according to the Euclidian distance and edges are draw between neighbors in the graph.
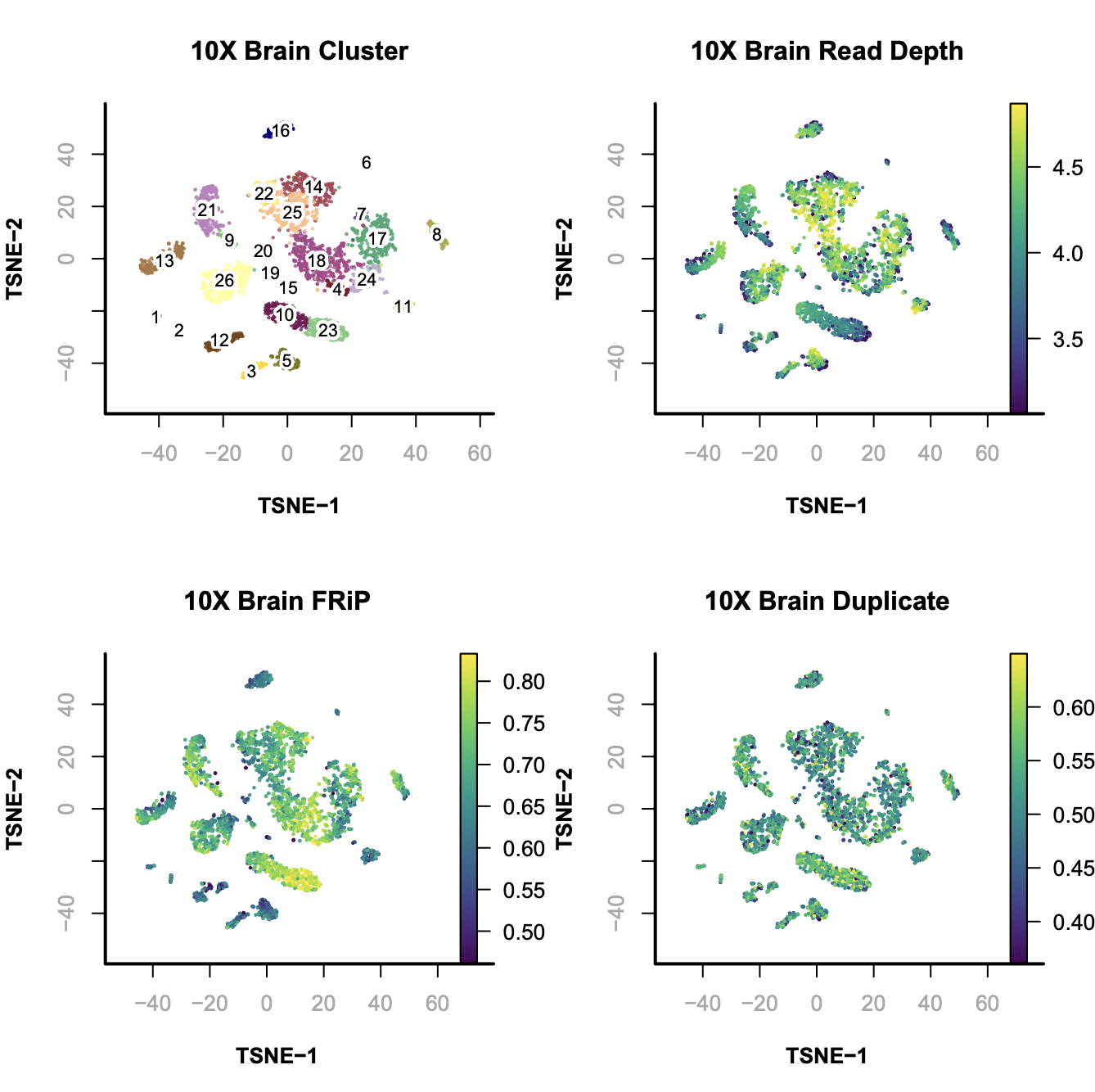
SnapATAC visualizes and explores the data using tSNE (FI-tsne) or UMAP. In this example, we compute the t-SNE embedding. We next project the sequencing depth or other bias onto the t-SNE embedding.
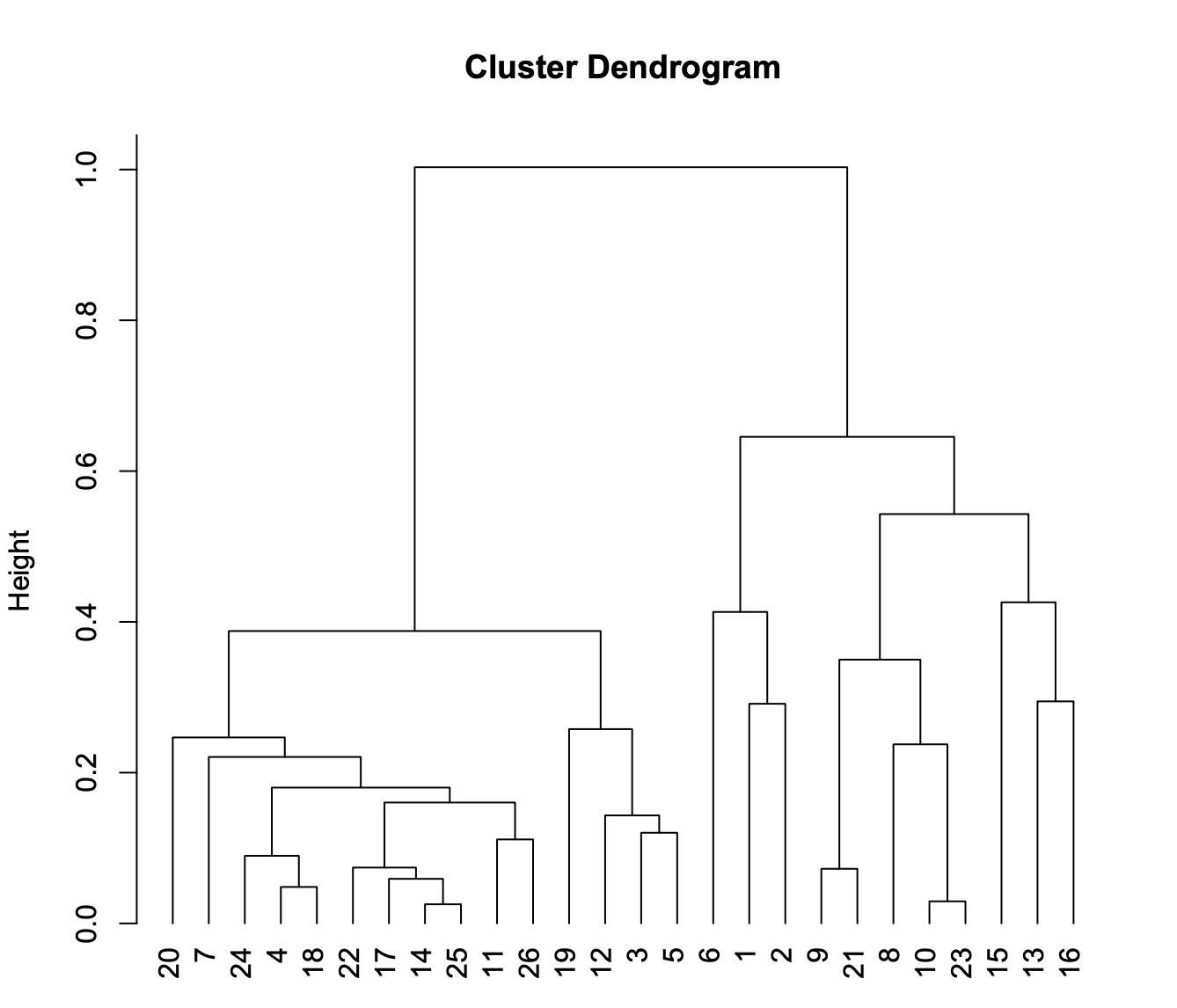
Next, cells belonging to the same cluster are pooled to create the aggregate signal for heretical clustering.
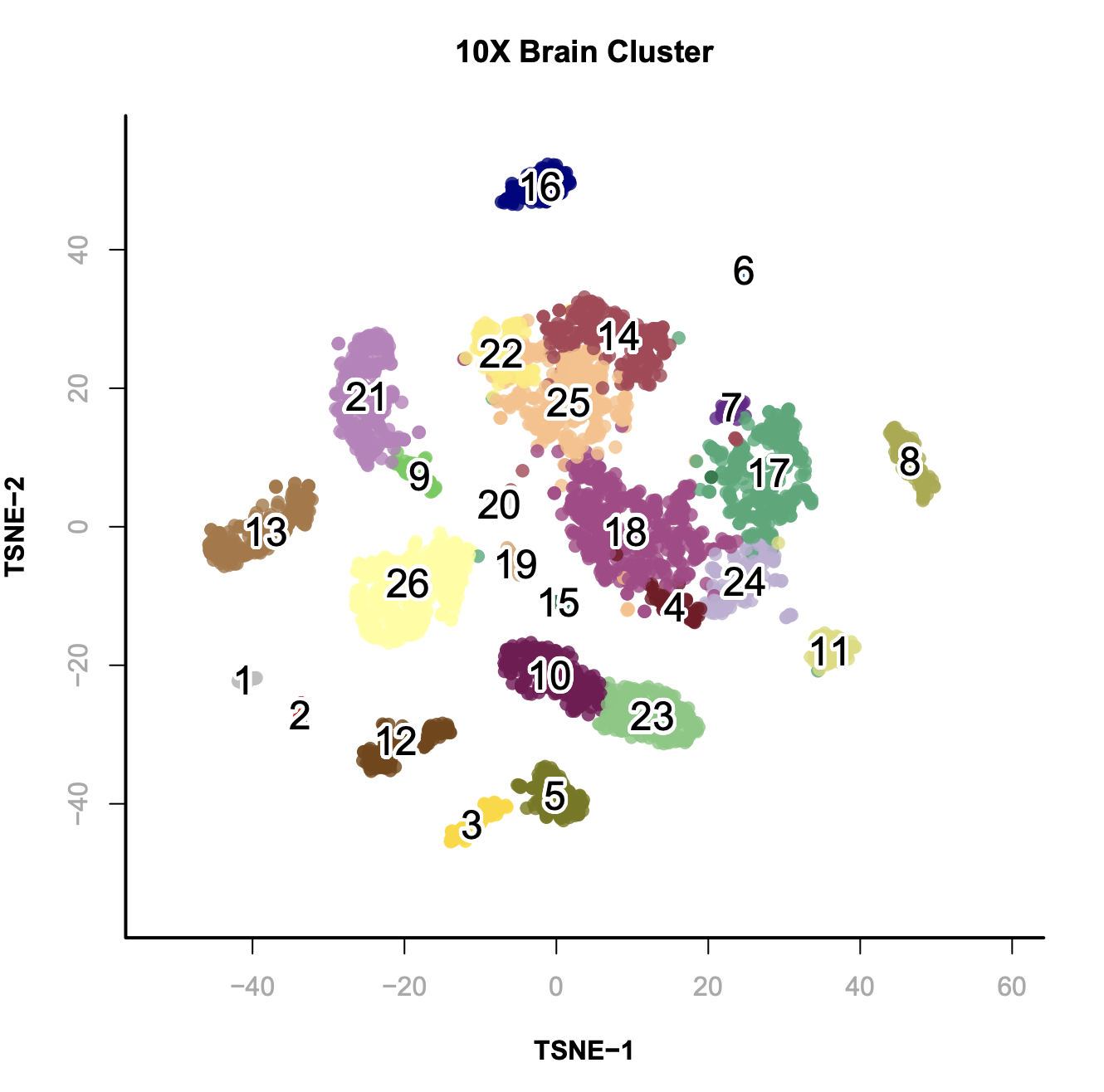
# calculate the ensemble signals for each cluster > ensemble.ls = lapply(split(seq(length(x.sp@cluster)), x.sp@cluster), function(x){ SnapATAC::colMeans(x.sp[x,], mat="bmat"); }) # cluster using 1-cor as distance > hc = hclust(as.dist(1 - cor(t(do.call(rbind, ensemble.ls)))), method="ward.D2"); > plotViz( obj=x.sp, method="tsne", main="10X Brain Cluster", point.color=x.sp@cluster, point.size=1, point.shape=19, point.alpha=0.8, text.add=TRUE, text.size=1.5, text.color="black", text.halo.add=TRUE, text.halo.color="white", text.halo.width=0.2, down.sample=10000, legend.add=FALSE ); > plot(hc, hang=-1, xlab="");
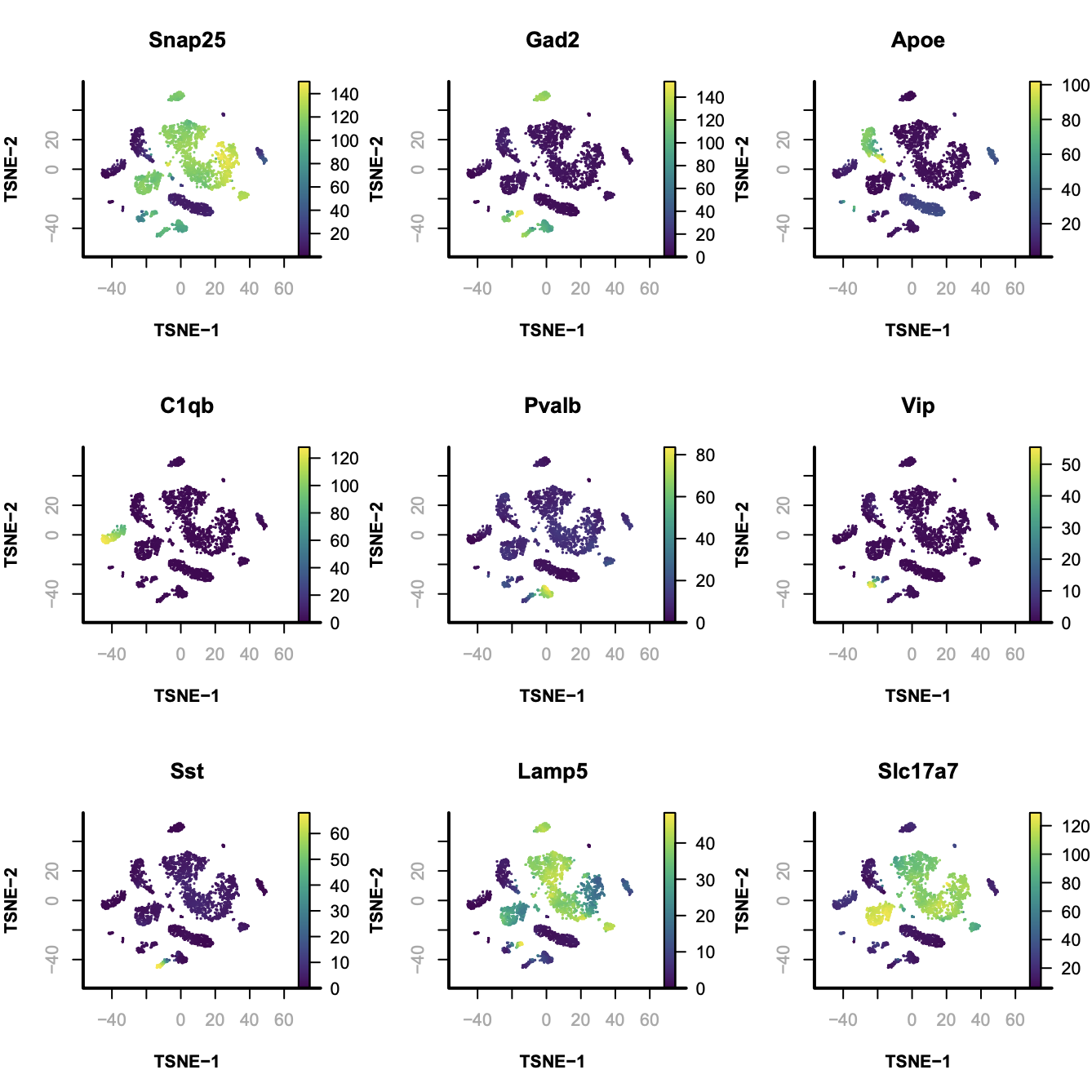
In this case, from cluster 20 to 25 are excitatory neuron; cluster 19 to 5 are inhibitory neurons and the rest of them are non-neuronal cells.
Step 11. Identify peaks
Next we aggregate cells from the each cluster to create an ensemble track for peak calling and visualization. This step will generate a .narrowPeak file that contains the identified peak and .bedGraph file for visualization. To obtain the most robust result, we don’t recommend to perform this step for clusters with cell number less than 100. In the below example, SnapATAC creates atac_v1_adult_brain_fresh_5k.1_peaks.narrowPeak and atac_v1_adult_brain_fresh_5k.1_treat_pileup.bdg. bdg file can be compressed to bigWig file using bedGraphToBigWig for IGV or Genome Browser visulization.
## number of barcodes: 4100 ## number of bins: 546206 ## number of genes: 16 ## number of peaks: 242847 ## number of motifs: 0
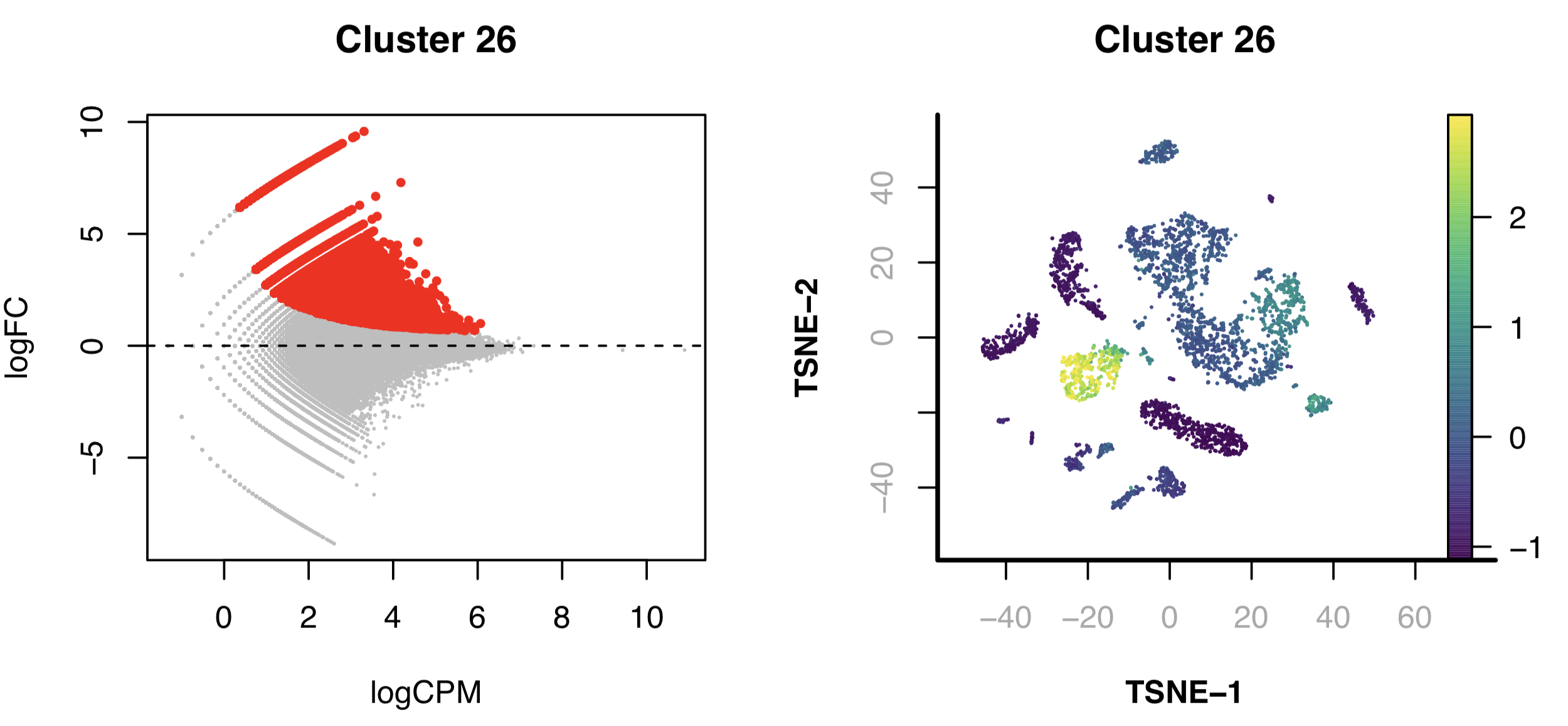
Step 14. Identify differentially accessible regions
SnapATAC finds differentially accessible regions (DARs) that define clusters via differential analysis. By default, it identifies positive peaks of a single cluster (specified in cluster.pos), compared to a group of negative control cells. If by default cluster.neg=NULL, findDAR will look for a group of background cells closest to the positive cells.
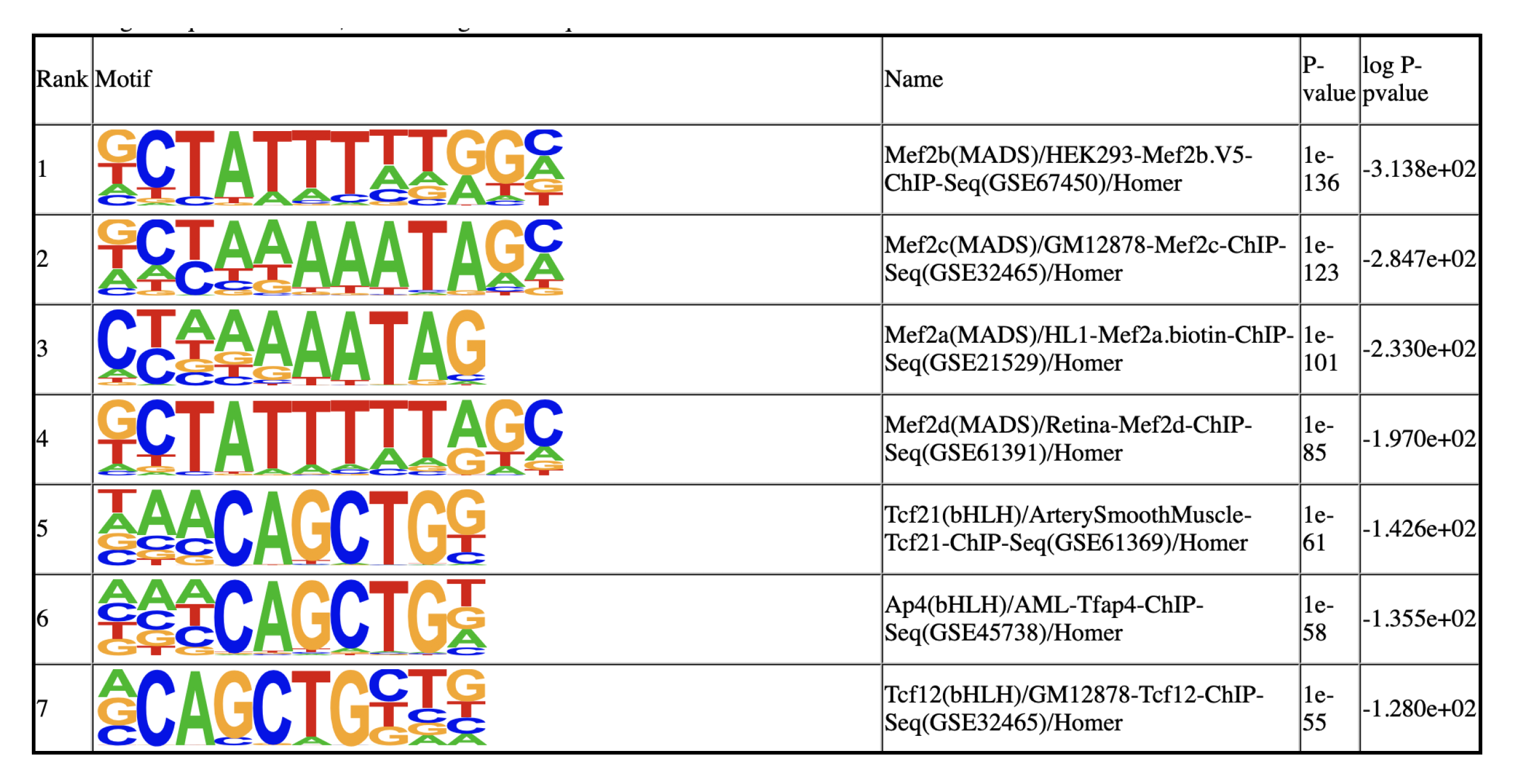
Next, we identify DARs for each of the clusters. For clusters, especially the small ones, that lack the static power to reveal DARs, we rank the peaks based on the enrichment and use the top 2000 peaks as representative peaks for motif discovery.
SnapATAC employs Homer to identify master regulators that are enriched in the differentially accessible regions (DARs). This will creates a homer motif report knownResults.html in the folder ./homer/C5. This requires Homer to be pre-installed. See here for the instruction about how to install Homer.
SnapATAC applies GREAT to identify biological pathways active in each of the cell cluster. In this example, we will first identify the differential elements in Microglia cells (cluster 13) and report the top 6 GO Ontologies enrichment inferred using GREAT analysis.
## install R package rGREAT > if (!requireNamespace("BiocManager", quietly=TRUE)) install.packages("BiocManager") > BiocManager::install("rGREAT") ## or install the latest version > library(devtools) > install_github("jokergoo/rGREAT")
## Submit time: 2019-09-0414:14:02 ## Version: default ## Species: mm10 ## Inputs: 25120 regions ## Background: wholeGenome ## Model: Basal plus extension ## Proximal: 5 kb upstream, 1 kb downstream, ## plus Distal: up to 1000 kb ## Include curated regulatory domains ## ## Enrichment tables for following ontologies have been downloaded: ## None
With job, we can now retrieve results from GREAT. The first and the primary results are the tables which contain enrichment statistics for the analysis. By default it will retrieve results from three GO Ontologies and all pathway ontologies. All tables contains statistics for all terms no matter they are significant or not. Users can then make filtering by a self-defined cutoff.
## ID name Binom_Genome_Fraction ## 1 GO:0002376 immune system process 0.12515840 ## 2 GO:0002682 regulation of immune system process 0.09012561 ## 3 GO:0009987 cellular process 0.80870120 ## 4 GO:0048518 positive regulation of biological process 0.43002240 ## 5 GO:0050789 regulation of biological process 0.68873070 ## 6 GO:0050794 regulation of cellular process 0.66837300 ## Binom_Expected Binom_Observed_Region_Hits ## 13095.9185592 ## 22229.3474148 ## 320004.03022241 ## 410637.03013697 ## 517036.44019871 ## 616532.87019356