Analysis of single-cell ATAC-seq data using SnapATAC software (3):Integrative Analysis of PBMC scATAC-seq and scRNA-seq
Posted onEdited onInscRNAViews: Symbols count in article: 20kReading time ≈19 mins.
Introduction
Integrative Analysis of PBMC scATAC-seq and scRNA-seq
In this example, we will be analyzing two scATAC-seq datasets (5K and 10K) and one scRNA-seq dataset from PBMC. All three datasets are freely available from 10X genomics.
In detail, we will be performing the following analysis:
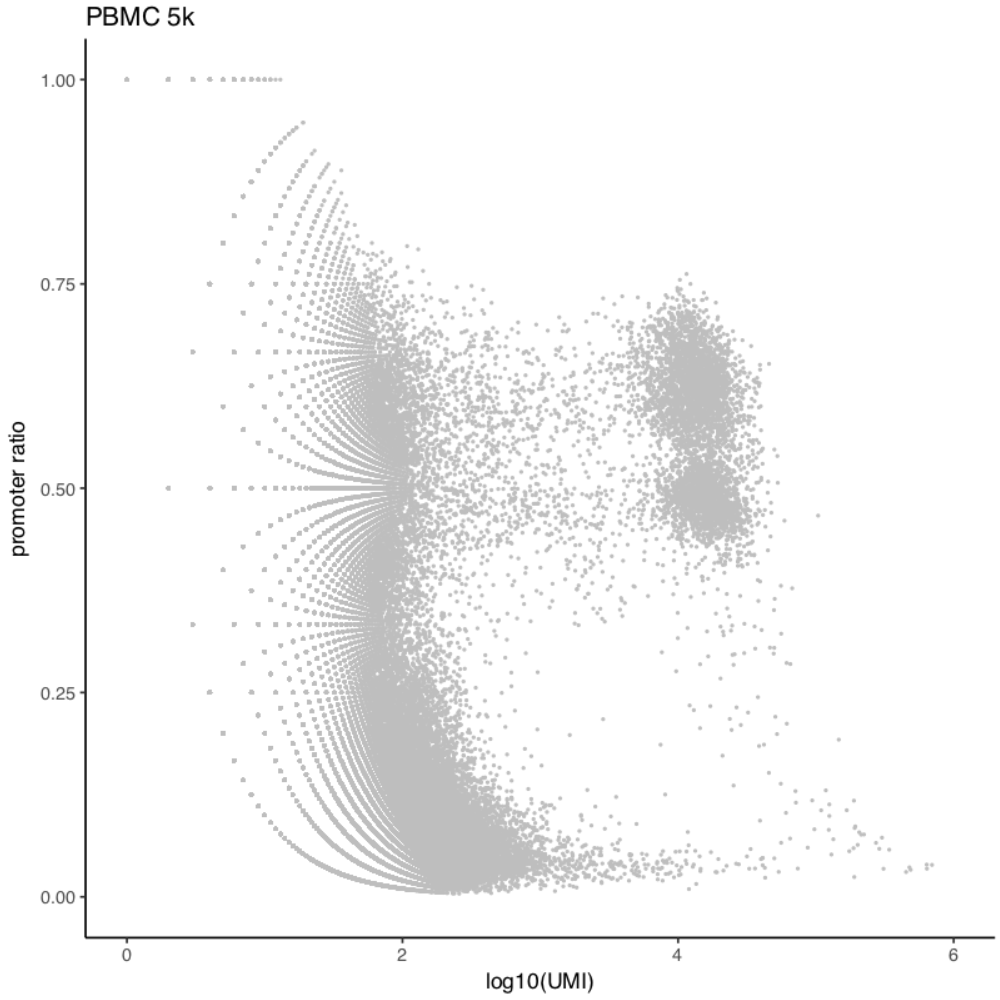
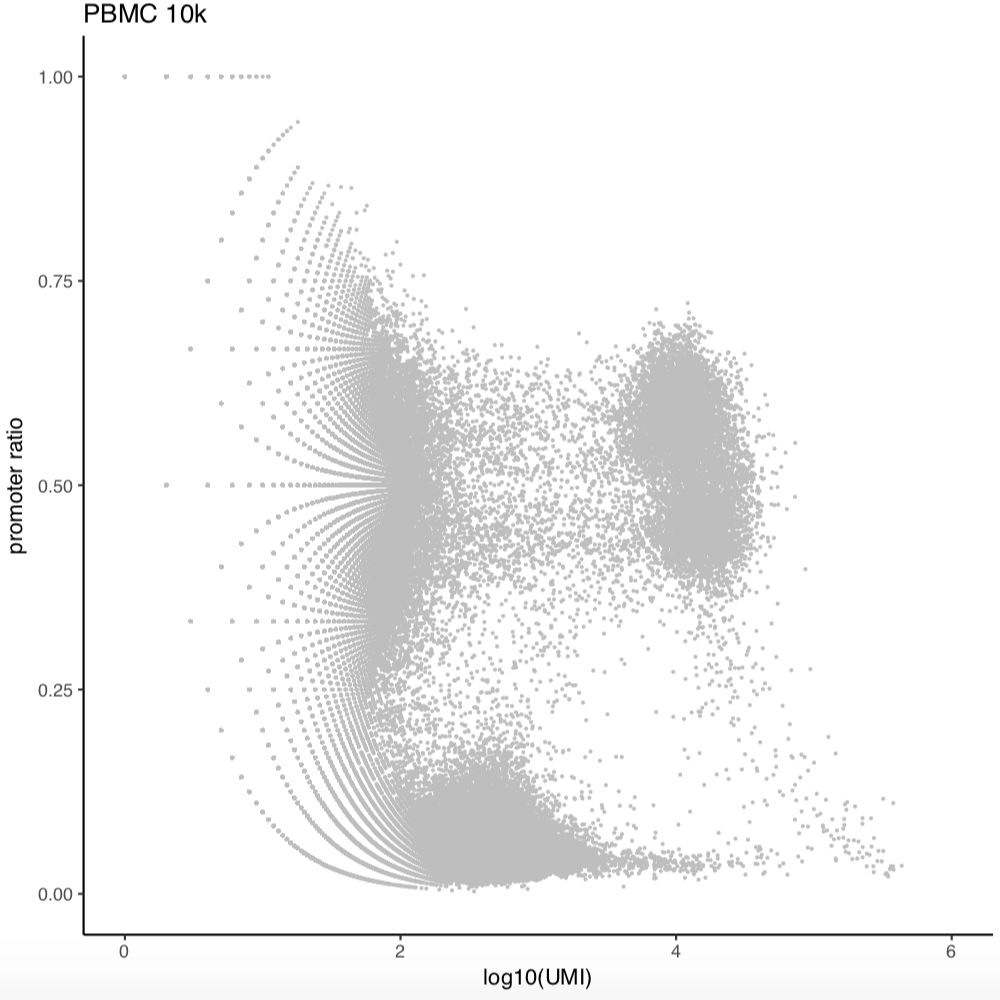
Cell selection for PBMC 5k and 10k scATAC;
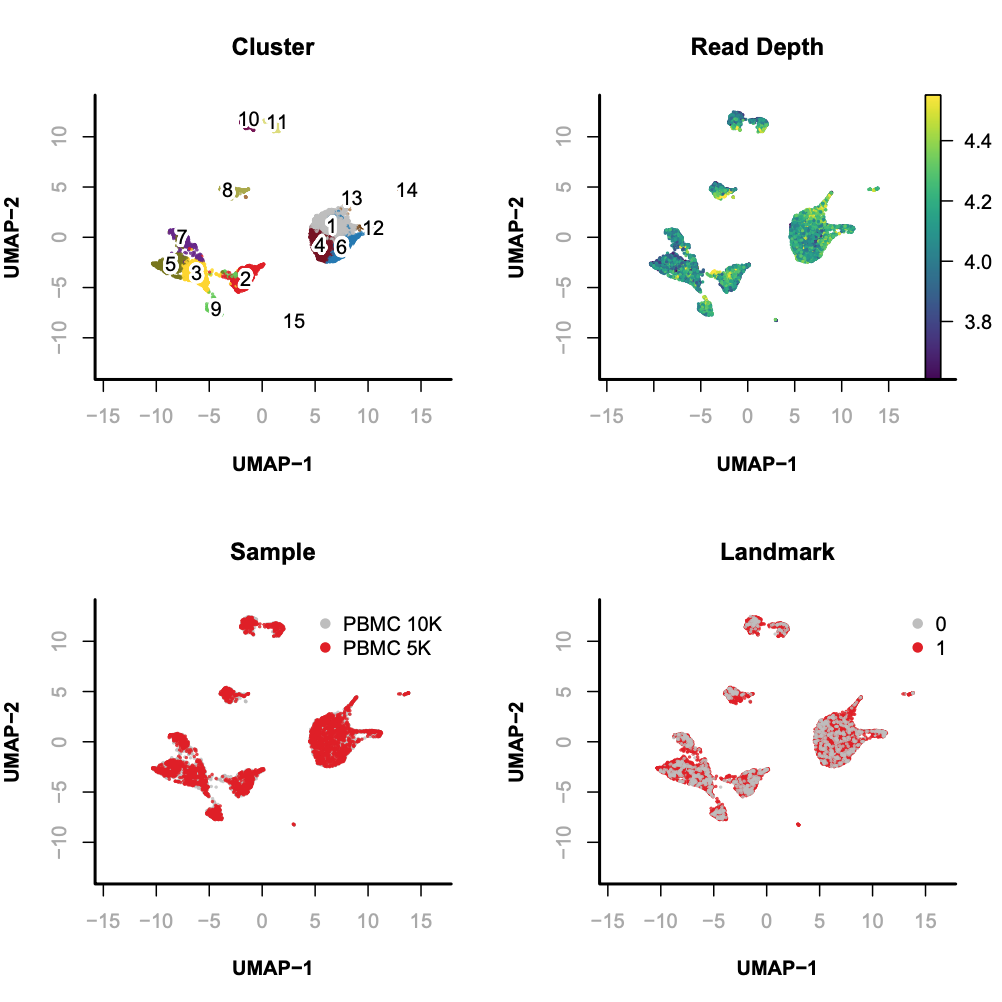
Randomly sample 10,000 cells as landmarks;
Unsupervised clustering of landmarks;
Project the remaining (query) cells onto the landmarks;
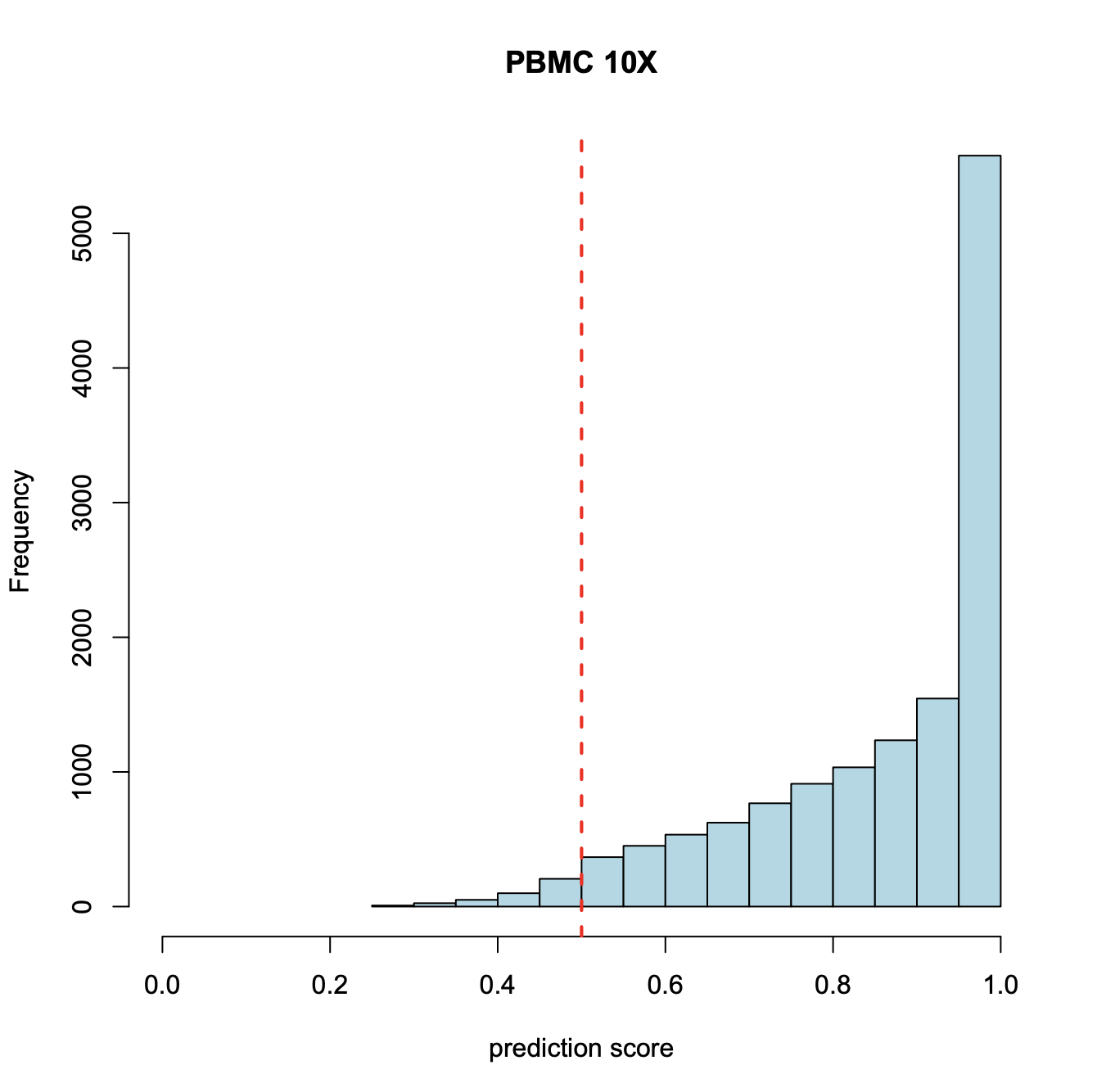
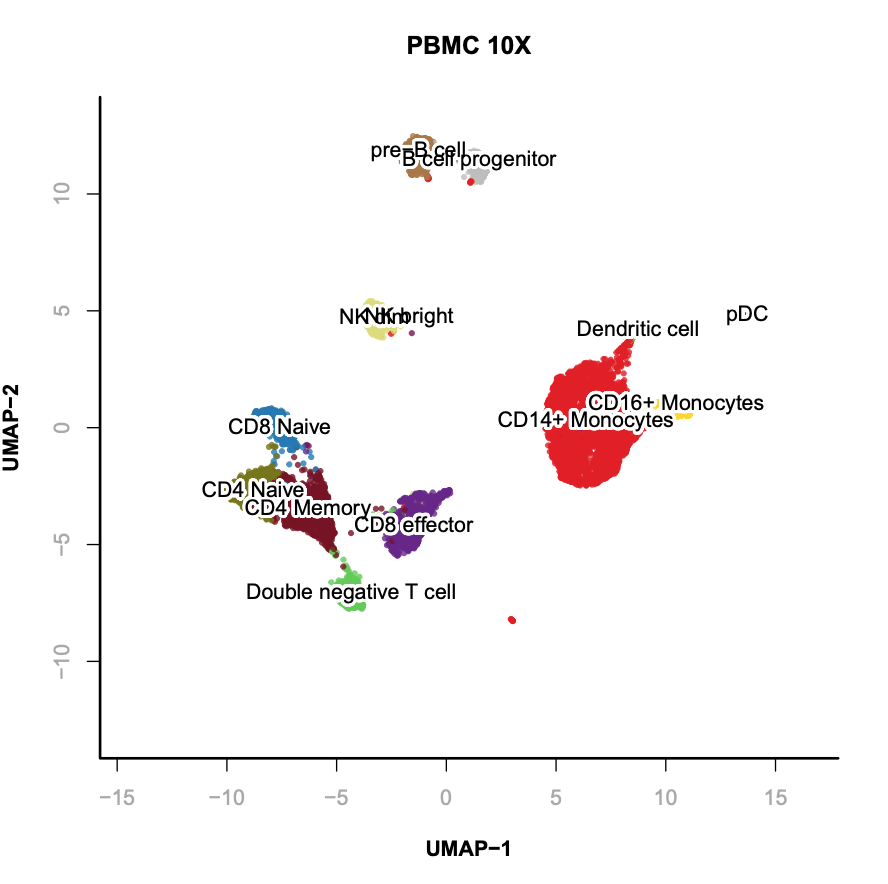
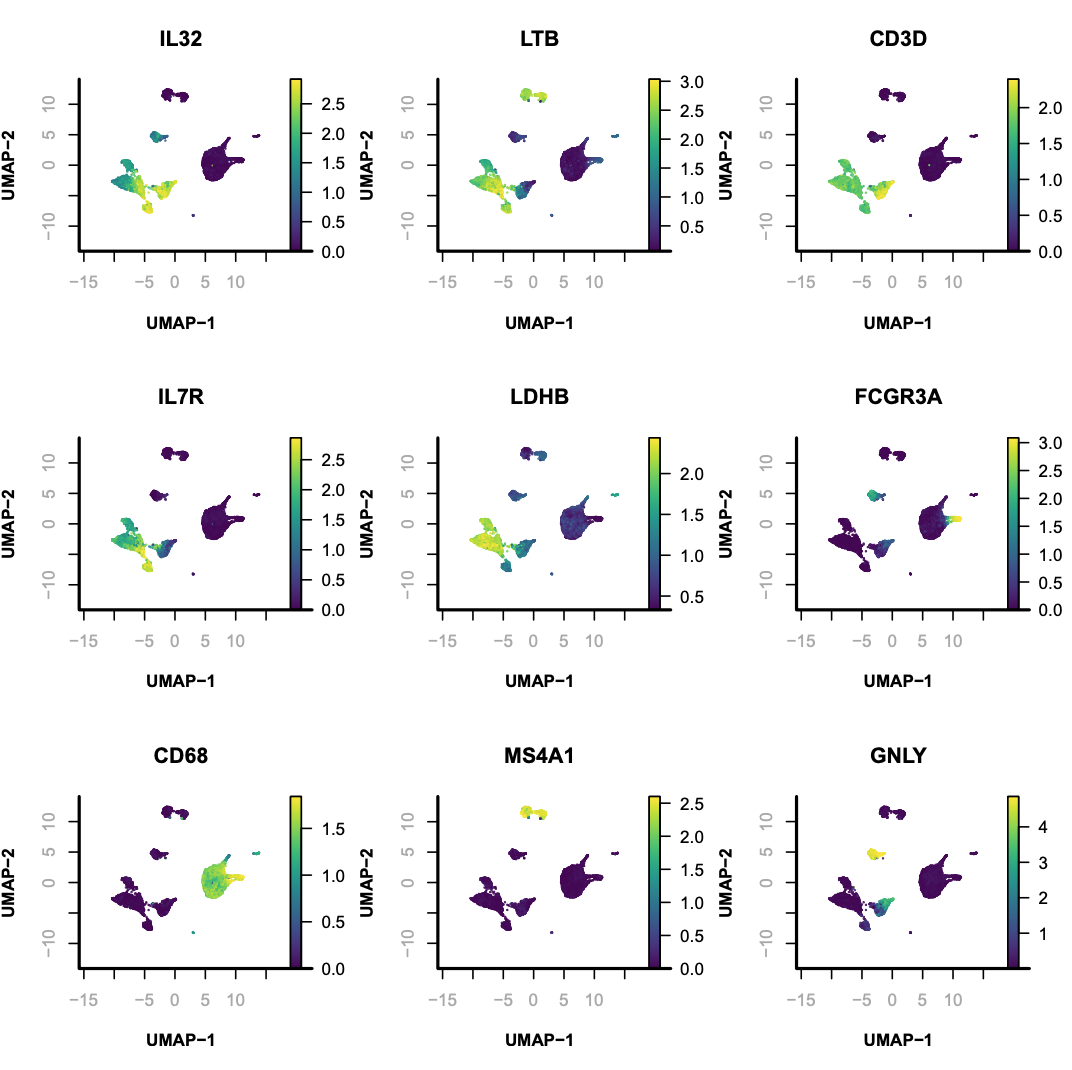
Supervised annotation of scATAC clusters using scRNA dataset;
Downstream analysis including peak calling, differential analysis, prediction of gene-enhancer pairing.
## $`PBMC 5K` ## number of barcodes: 20000 ## number of bins: 0 ## number of genes: 0 ## number of peaks: 0 ## number of motifs: 0 ## ## $`PBMC 10K` ## number of barcodes: 20000 ## number of bins: 0 ## number of genes: 0 ## number of peaks: 0 ## number of motifs: 0
## $`PBMC 5K` ## number of barcodes: 4526 ## number of bins: 0 ## number of genes: 0 ## number of peaks: 0 ## number of motifs: 0 ## ## $`PBMC 10K` ## number of barcodes: 9039 ## number of bins: 0 ## number of genes: 0 ## number of peaks: 0 ## number of motifs: 0
## number of barcodes: 13565 ## number of bins: 0 ## number of genes: 0 ## number of peaks: 0
> table(x.sp@sample);
## PBMC 10K PBMC 5K ## 90394526
Step 2. Add cell-by-bin matrix
Next, we add the cell-by-bin matrix of 5kb resolution to the snap object. This function will automatically read the cell-by-bin matrix from two snap files and add the concatenated matrix to bmat attribute of snap object.
> x.sp = addBmatToSnap(x.sp, bin.size=5000);
Step 3. Matrix binarization
We will convert the cell-by-bin count matrix to a binary matrix. Some items in the count matrix have abnormally high coverage perhaps due to the alignment errors. Therefore, we next remove top 0.1% items in the count matrix and then convert the remaining non-zero values to 1.
> x.sp = makeBinary(x.sp, mat="bmat");
Step 4. Bin filtering
First, we filter out any bins overlapping with the ENCODE blacklist to prevent from potential artifacts.
## number of barcodes: 13565 ## number of bins: 624297 ## number of genes: 0 ## number of peaks: 0 ## number of motifs: 0
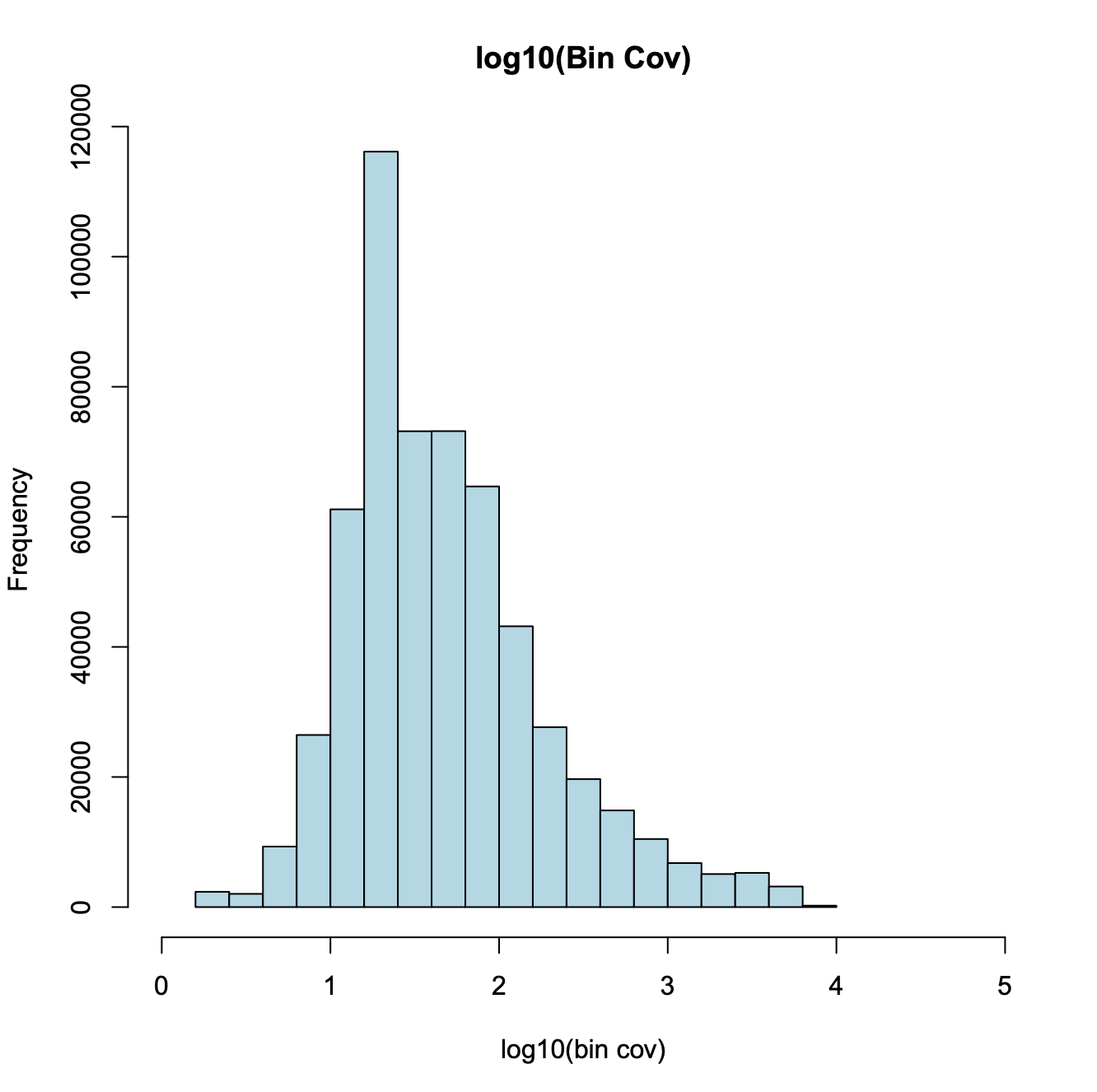
Third, the coverage of bins roughly obeys a log normal distribution. We remove the top 5% bins that overlap with invariant features such as the house keeping gene promoters.
## number of barcodes: 13565 ## number of bins: 534985 ## number of genes: 0 ## number of peaks: 0 ## number of motifs: 0
Next, we will further remove any cells of bin coverage less than 1,000. The rational behind this is that some cells may have high number of unique fragments but end up with low bin coverage after filtering. This step is optional but highly recommanded.
## number of barcodes: 13434 ## number of bins: 534985 ## number of genes: 0 ## number of peaks: 0 ## number of motifs: 0
Step 5. Dimentionality reduction
SnapATAC applies diffusion maps algorithm, a nonlinear dimensionality reduction technique that discovers low-dimension manifold by performing random walk on the data and is highly robust to noise and perturbation.
However, the computational cost of the diffusion maps algorithm scales exponentially with the increase of number of cells. To overcome this limitation, here we combine the Nyström method (a sampling technique) and diffusion maps to present Nyström Landmark diffusion map to generate the low-dimentional embedding for large-scale dataset.
A Nyström landmark diffusion maps algorithm includes three major steps:
***sampling***: sample a subset of K (K≪N) cells from N total cells as “landmarks”. Instead of random sampling, here we adopted a density-based sampling approach developed in SCTransform to preserve the density distribution of the N original points;
***embedding***: compute a diffusion map embedding for K landmarks;
***extension***: project the remaining N-K cells onto the low-dimensional embedding as learned from the landmarks to create a joint embedding space for all cells.
In this example, we will sample 10,000 cells as landmarks and project the remaining query cells onto the diffusion maps embedding of landmarks.
Combine landmark and query cells. Note: To merge snap objects, all the matrix (bmat, gmat, pmat) and metaData must be of the same number of columns between snap objects.
Step 6. Determine significant components
We next determine the number of eigen-vectors to include for downstream analysis. We use an ad hoc method by simply looking at a pairwise plot and select the number of eigen vectors that the scatter plot starts looking like a blob. In the below example, we choose the first 15 eigen vectors.
Step 7. Graph-based clustering
Using the selected significant components, we next construct a K Nearest Neighbor (KNN) Graph. Each cell is a node and the k-nearest neighbors of each cell are identified according to the Euclidian distance and edges are draw between neighbors in the graph.
Step 10. Create psudo multiomics cells
Now each single cell in the snap object x.sp contains information of both chromatin accessibility @bmat and gene expression @gmat.
Step 13. Identify peaks
Next we aggregate reads from the each cluster to create an ensemble track for peak calling and visualization. This step will generate a narrowPeak that contains the identified peak and .bedGraph file for visualization. To obtain the most robust result, we don’t recommend to perform this step for clusters with cell number less than 100. In the below example, SnapATAC creates PBMC.CD4_Naive_peaks.narrowPeak and PBMC.CD4_Naive_treat_pileup.bdg. bdg file can be compressed to bigWig file using bedGraphToBigWig for IGV or Genome Browser visulization.
Instead of performing peak calling for only one cluster, we provide a short script that perform this step for all clusters.
# call peaks for all cluster with more than 100 cells > clusters.sel = names(table(x.sp@cluster))[which(table(x.sp@cluster) > 100)]; > peaks.ls = mclapply(seq(clusters.sel), function(i){ print(clusters.sel[i]); peaks = runMACS( obj=x.sp[which(x.sp@cluster==clusters.sel[i]),], output.prefix=paste0("PBMC.", gsub(" ", "_", clusters.sel)[i]), path.to.snaptools="/home/r3fang/anaconda2/bin/snaptools", path.to.macs="/home/r3fang/anaconda2/bin/macs2", gsize="hs", # mm, hs, etc buffer.size=500, num.cores=1, macs.options="--nomodel --shift 100 --ext 200 --qval 5e-2 -B --SPMR", tmp.folder=tempdir() ); peaks }, mc.cores=5); > peaks.names = system("ls | grep narrowPeak", intern=TRUE); > peak.gr.ls = lapply(peaks.names, function(x){ peak.df = read.table(x) GRanges(peak.df[,1], IRanges(peak.df[,2], peak.df[,3])) }) > peak.gr = reduce(Reduce(c, peak.gr.ls));
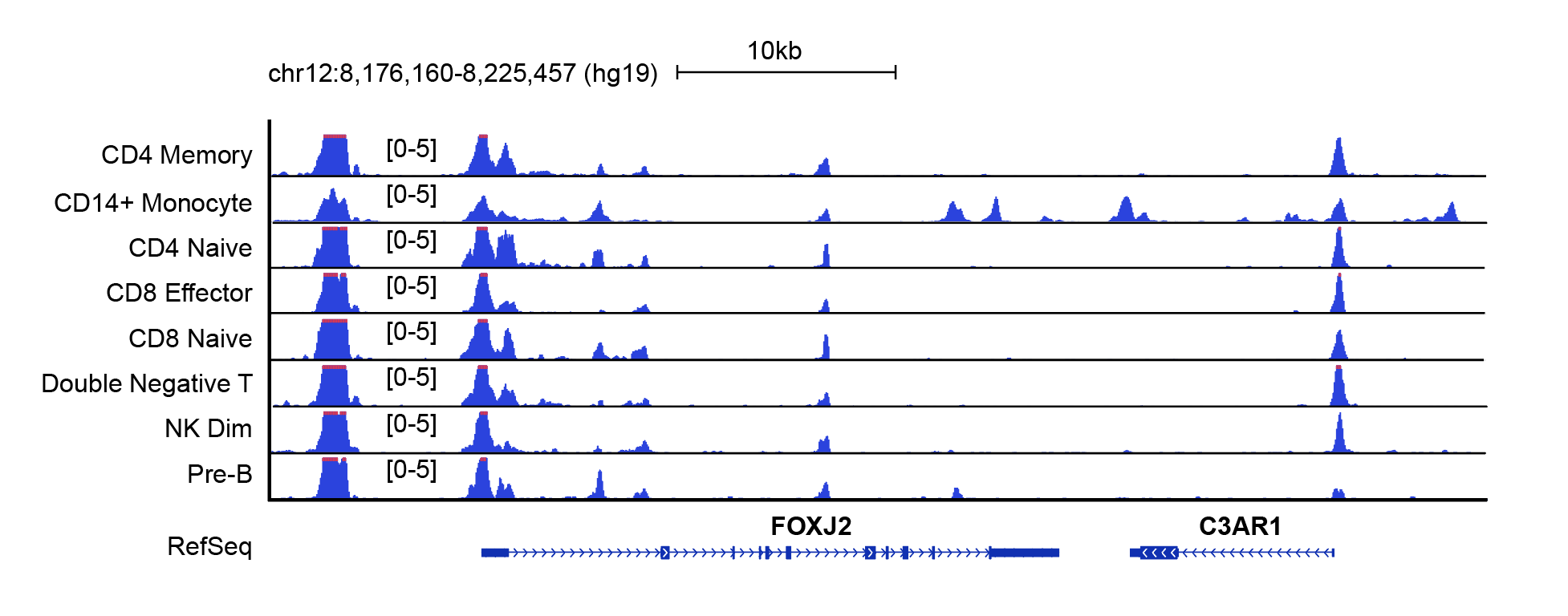
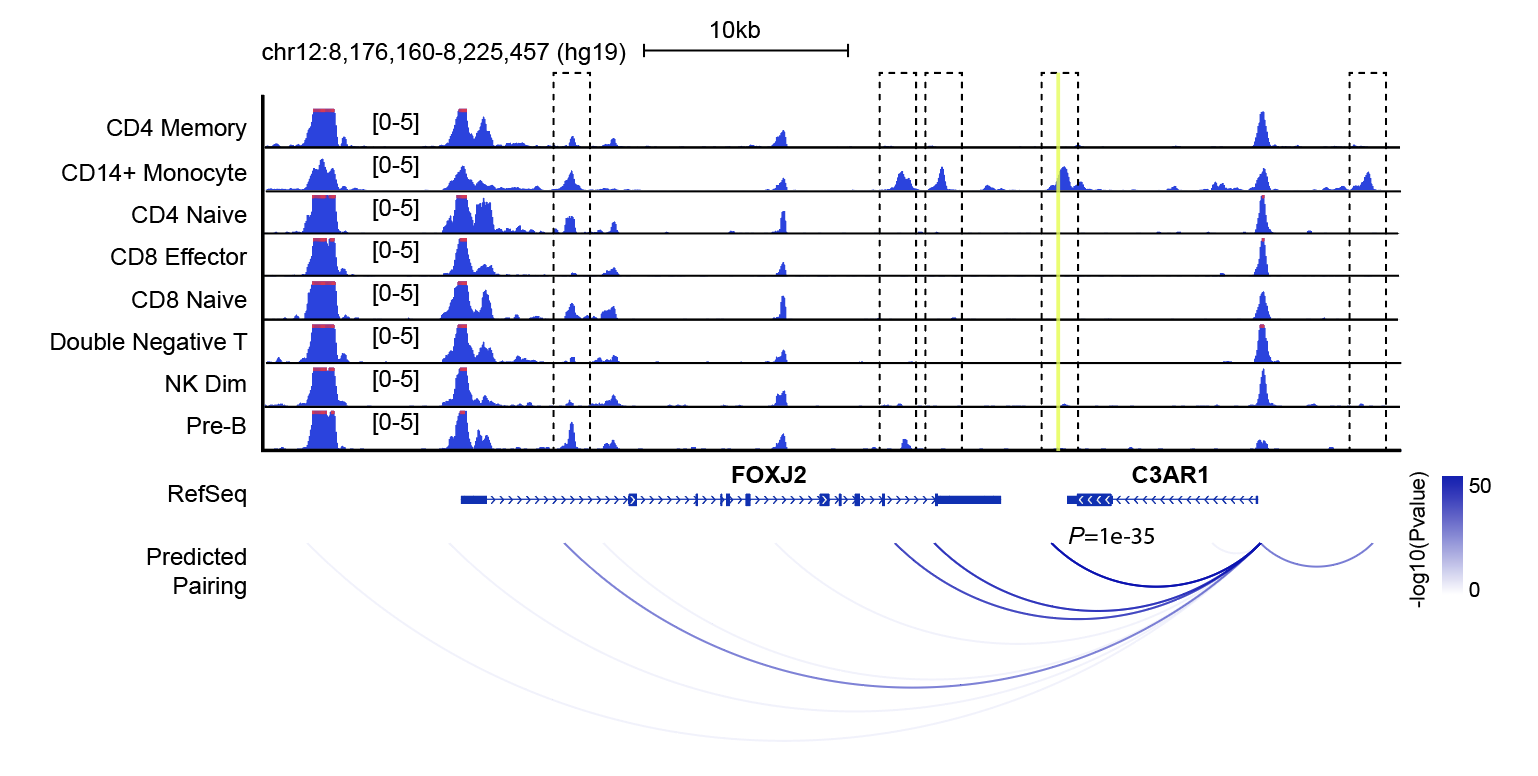
This will create a bdg file for each cluster for visilizations using IGV or other genomic browsers such as UW genome browser. Below is a screenshot of regions flanking FOXJ2 gene from UW genome browser.
Step 14. Create a cell-by-peak matrix
Using merged peaks as a reference, we next create the cell-by-peak matrix and add it to the snap object. We will first write down combined peak list as peaks.combined.bed.
We then add the cell-by-peak matrix to the existing snap object in R.
> x.sp = addPmatToSnap(x.sp);
Step 15. Identify differentially accessible peaks
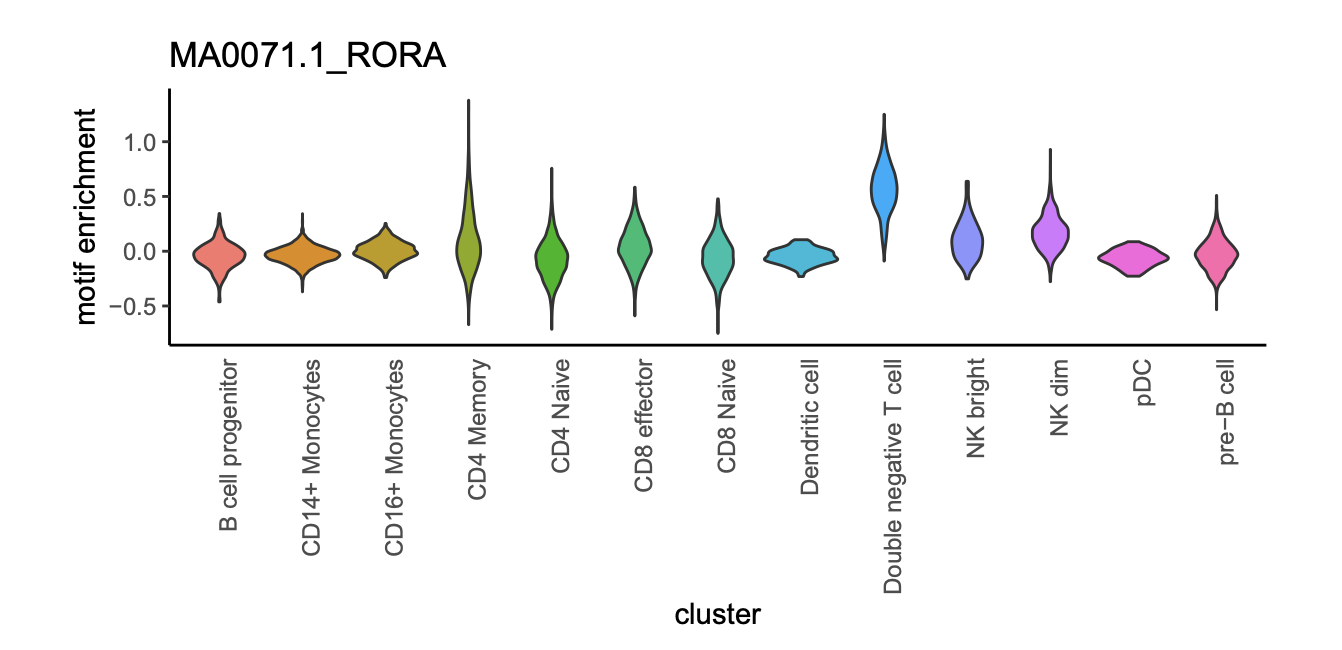
For a given group of cells Ci, we first look for their neighboring cells Cj (|Ci|=|Cj|) in the diffusion component space as “background” cells to compare to. If Ci accounts for more than half of the total cells, we use the remaining cells as local background. Next, we aggregate Ci and Cj to create two raw-count vectors as Vci and Vcj. We then perform differential analysis between Vci and Vcj using exact test as implemented in R package edgeR (v3.18.1) with BCV=0.1 for mouse and 0.4 for human. P-value is then adjusted into False Discovery Rate (FDR) using Benjamini-Hochberg correction. Peaks with FDR less than 0.05 are selected as significant DARs.
We recognize the limitation of this approach is that the statically significance may be under power for small clusters. For clusters that failed to identify significant differential elements, we rank the elements based on the enrichment pvalue and pick the top 2,000 peaks a representative elements for motif analysis.
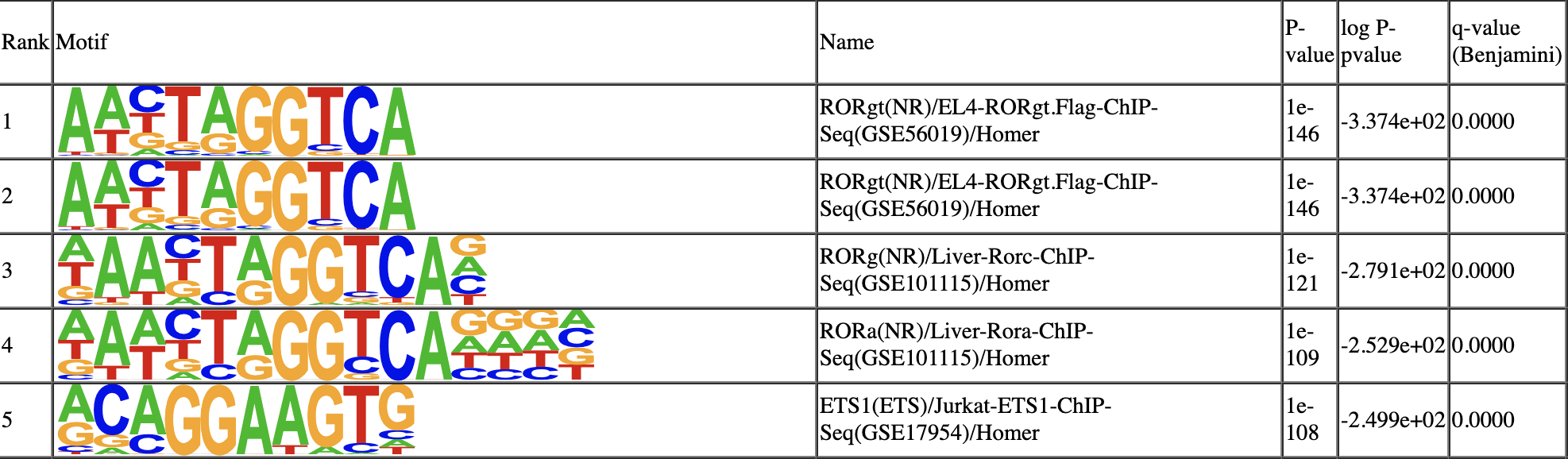
Step 17. De novo motif discovery
SnapATAC can help identify master regulators that are enriched in the differentially accessible regions (DARs). This will creates a homer motif report knownResults.html in the folder ./homer/C2.
Step 18. Predict gene-enhancer pairs
Finally, using the “pseudo” cells, we next develop a method to link the distal regulatory elements to the target genes based on the association between expression of a gene and chromatin accessibility at its distal elements in single cells. For a given marker gene, we first identify peaks within a flanking window the target gene. For each flanking peak, we perform logistic regression using gene expression as input varaible to predict the binarized chromatin state. The resulting model estimates the significance of the association between chromatin accessibility and gene expression.